This work is conducted in collaboration with Prof. Sharon H. Ackerman.
The stability landscape of KDO8PS.
From the point of view of protein stability the organization of enzyme active sites is inherently unstable because these sites are optimized for catalysis, which means they are pre-organized to stabilize the transition state(s), rather than the protein. Thus, the substitution of a catalytic side chain (most often to alanine) will typically increase the overall protein stability, while sacrificing function. Conversely, most mutations that introduce new function are destabilizing. The generality of this stability-function tradeoff must be viewed within the context of the fact that regardless of their effect on functions most mutations are destabilizing. With respect to KDO8PS, several attempts were made to map the evolutionary paths from metallo- to non-metallo forms (and vice versa) without giving sufficient consideration to the fact that sequence differences between and within these forms in different bacterial backgrounds, may be related not only to activity but also to stability.
One way we looked at the relevance of individual residues in the context of the three-dimensional structure of KDO8PSs was by computing a Hidden Markov Model (HMM) for the protein family represented by the combined metallo (175 sequences with Cys at position 23 of Neisseria meningitidis (Nm.) serogroup B KDO8PS (Uniref Q9JZ55, PDB 2QKF), taken here as the reference sequence, the ‘C23’ sub-family) and non-metallo (173 sequences with Asn at position 23, the ‘N23’ sub-family) proteins . A HMM profile specifies a probability distribution over the alphabet of the 20 common amino acids, taking into consideration the background frequency for each amino acid, computed by counting amino acid occurrences in all known proteins, or only in the proteins of the family under consideration. If the background frequency of amino acid j is pj, then the important positions are those whose distribution differs from p. Therefore, the relative entropy P between the observed distribution i iat the -th position of the profile and p, H(Pi || p) = I(Pi), defines the information content of Pi. This information can be visualized as a HMM histogram or vector. In practice, the height (relative entropy = information content) of the histogram at each position indicates how much the observed frequency of amino acids at that position deviates from the background frequency.
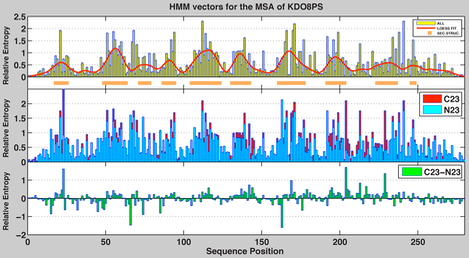
In the HMM vector, position numbers refer to the reference sequence of Nm. KDO8PS. There is a “wavy” pattern in the histogram derived from the MSA of all KDO8PSs (1st inset of the figure), which is clearly evident in the superimposed Loess fit. Regions of the histogram higher than the mean value are marked with an orange bar below the sequence; these regions invariably correspond to the strands of the core b-barrel in the structure, a clear indication that the sequence of the interior of the protein is less determined by chance. The HMM vectors of metallo (C23) or non-metallo (N23) KDO8PSs, calculated independently, are shown in the 2nd inset . Their difference is shown in the 3rd inset . The difference vector identifies positions that best fulfill the specific functional demands of the two subfamilies of KDO8PS. As expected, these positions tend to cluster outside the beta-barrel, which is the shared structural core of the entire family.
Overall, the HMM vectors of KDO8PSs suggest that evolution is primarily acting by preserving the structure of the protein interior. However, the vectors do not reveal whether the driving force for this effect is only function or whether stability is also a factor. This question was addressed by using the experimentally validated FoldX algorithm to calculate the DDG changes associated with introducing any one of the 20 possible amino acids at each position of the structure of Nm. KDO8PS (again taken here as the reference structure for this class of proteins). In our case we used an entire tetramer of KDO8PS, which is known to be the biological unit of the enzyme, and the individual mutations were introduced simultaneously in all four subunits. Thus, the calculated DDG changes account also for the effects of mutations at the interface between subunits.
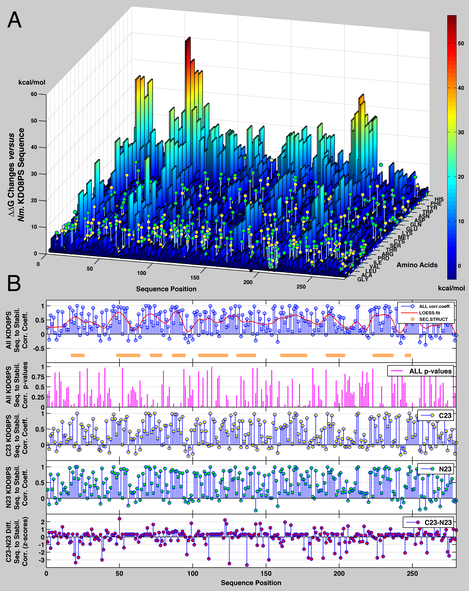
The outcome of this calculation is a “stability landscape” of KDO8PS: the peaks in the landscape represent positions in the protein where introduction of a certain amino acid would significantly increase the DG, and therefore decrease the overall stability. It is worth noting at this point that while the energies derived from FoldX are clearly not on an absolute scale, the relative trends are expected to be correct. In general, it can be seen how bulky aromatic residues (W,Y,F,H) tend to decrease stability ( = increase energy) at every position, and in four positions (21,68,231,232) any residue besides glycine or alanine decreases stability dramatically. These effects appear to be due to very large energy terms derived from van der Waals clashes of these residues with their surroundings, which are not sufficiently relieved by the relaxation of the structure. The observed amino acid frequencies at each position in the C23 and N23 sub-families of KDO8PSs are shown as lolly-pops (yellow for C23, green for N23, height proportional to the corresponding frequency) superimposed to the stability landscape of KDO8PS. In most positions, the amino acids most often used are those that do not decrease stability: in other words, only the planes of the stability landscape are significantly populated.
These data offer an interesting opportunity to understand the relationship between sequence and stability in KDO8PS: for each position in the reference structure of Nm. KDO8PS we consider two vectors (each with 20 elements): the first is the vector of the DDG changes associated with mutating the original sequence to each of the 20 amino acids; the other contains the frequencies of each of the 20 amino acids for that position in each of the two subfamilies of KDO8PS (or in the entire family). We recall that if a MSA is composed of independent sequences all producing stable folds with approximately the same structure, and if individual residues contribute additively to stability (no epistasis), then the stability contribution DDGa,i of a particular amino acid a at a given position i should be a roughly logarithmic function of its frequency fa,i in the MSA:
![]()
Approaches based on this idea have been generally successful in engineering more stable proteins. It follows that for each of the 280 positions in the reference structure of Nm. KDO8PS we can calculate the linear correlation coefficient (corr) between the stability vector [exp(-DDGa,i)] derived with FoldX and the amino acid frequency vector derived from the MSA. The result of this calculation is shown in the first four insets of Figure 2B; in the family of all KDO8PSs and in both subfamilies there is a clear distinction between positions in which the amino acid usage is influenced by an evolutionary drive to increase stability, and other positions in which there is no such trend. There is a wavy pattern in the correlation between stability and observed amino acid frequencies (see the Loess fit in the first inset), which is similar to that already noticed in the HMM vector. The 2nd inset of Figure 2B shows the p-values for the correlations in the 1st inset, calculated by testing the null hypothesis of zero correlation against the alternative hypothesis of non-zero correlation. The large p-values for all the sequence positions with correlation near zero, and the small p-values (<5E-4) for all the sequence positions with large correlation, lend credibility to the apparent correlation between stability gains and choice of particular amino acids in certain regions of the protein. The correlation coefficients between the HMM vectors of relative entropies and the stability/sequence correlation vectors are 0.34 (p=7.1E-9), 0.31 (p=1.5E-7), and 0.39 (p=1.9E-11), for all KDO8PSs and for the C23 and N23 subfamilies, respectively, suggesting that the choice of sequence in the core beta-barrel is at least in part aimed at increasing the overall stability of the structure. If the correlation vectors between sequence and stability for each sub-family (3rd and 4th insets) are subtracted from each other (5th inset), a new set of positions is identified in the structure of Nm. KDO8PS, which represent places in which the correlation with stability changes significantly between the two sub-families. These are positions in which one sub-family selectively affects stability with respect to the other sub-family by preferentially adopting or discarding certain amino acids. By and large, these positions are also found outside the shared beta-barrel core of both sub-families, as already observed for the difference between the HMM vectors.
The contribution of coevolving residues to stability in KDO8PS.
In a MSA some positions are highly conserved, while others vary. The conserved positions are clearly important, but the non-conserved positions are not irrelevant because the net stabilization of the folded state, relative to the unfolded state is usually so small, that all positions may contribute significantly to the protein stability. This is clearly evident in the stability landscape of KDO8PS. Thus, the destabilizing effects of a given amino acid at one position can be compensated by the stabilizing effect of a certain amino acid at another position: in other words, two positions could be coevolving. A wide variety of algorithms have been developed to detect coevolving positions from a MSA (see MSAVOLVE page). Some of these methods work within the frame of information theory [49]. Information entropy, H(X), is a measure of the uncertainty associated with a discrete random variable X that assumes values {x1, ..., xn}:
![]()
where b is the base of the logarithm used and p is the probability mass function of the variable X [50,51]. Related to H(X), mutual information, MI(X;Y), measures the mutual dependence of two discrete random variables X and Y:
![]()
where p(x,y) is the joint probability mass function of X and Y, and p(x) and p(y) are the marginal probability mass functions of X and Y, respectively. Intuitively, MI measures how much knowing one of the two variables reduces the uncertainty about the other. In a MSA, the amino acids in a given column can be considered as a set of observations (xi) of a random variable X. An estimate of the entropy H(X) is obtained by using the observed amino acid frequencies, f(xi), in place of the underlying probabilities, p(xi); likewise, MI(X;Y) for a pair of columns can be derived using the frequencies, f(xi,yj), of all ordered pairs occurring in the two columns. In practice, MI between positions (columns in a MSA) reflects the extent to which knowledge of the amino acid at one position allows us to predict the identity of the amino acid at the other position. If amino acids occur independently at the two sites, the theoretical value for MI is zero; conversely, MI is high if the two positions are correlated.
However, significant background MI can originate from random pairings of residues when the number of sequences in the multiple sequence alignments is small (in practice, less than 125 sequences [54]). In addition, positions with high entropy (non-conserved positions) have more background MI than positions with low entropy. MI is also affected by various sources of bias, because the sequences in a MSA do not exactly meet the assumption of independent evolution. For example, the appearance of a mutation in an ancestral protein, which is clearly a single evolutionary event, would be considered in a MI analysis as representing an independent event that occurred in each of the proteins in the MSA that descended from that ancestor. This treatment of a single event as multiple independent events acts as a phylogenetic bias that increases the mutual information among residues. Normalization of MI values reduces the effect of positional entropy and phylogenetic bias, and several normalized variants of MI have been proposed. including MIp [58], Z-scored residual MI, ZRes, Z-scored-product normalized MI, ZNMI. The Zpx score introduced by Gloor supercedes the MIp score.
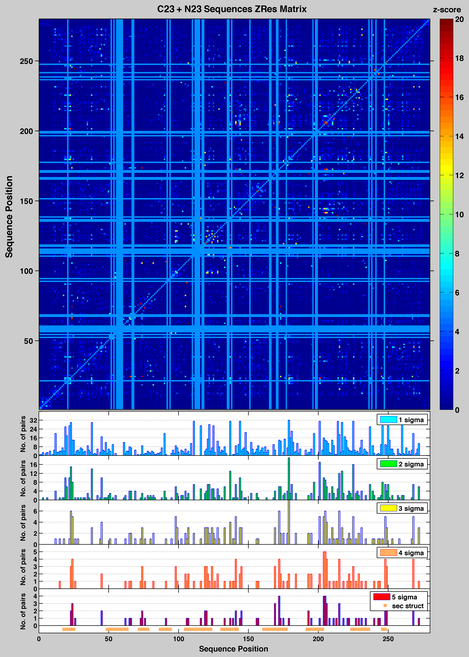
The Zpx, ZRes and ZNMI matrices all work well with the KDO8PS data set in reducing the background MI; as expected they are highly correlated to each other (corr(Zpx,ZRes) = 0.90, corr(Zpx,ZNMI) = 0.81, corr(ZNMI,ZRes) = 0.73). In this study we were not interested in determining whether there are patterns of spatial relationship among the coevolving positions, but whether there is a relationship between coevolving pairs and the stability landscape of KDO8PS described in the previous section. As an example of our approach, the number of coevolving pairs with score higher than 1 to 5 s over the mean of all scores in the ZRes matrix, are shown for every position in the MSA of all KDO8PSs as histograms in the insets of the figure on the side. The same regions of sequence highlighted for the HMM vector are shown below the baseline of the lowest inset, as horizontal orange bars.
Since the HMM vector represents deviations from the expected background probability distribution to satisfy the demands of structure, stability and function, it is reasonable to suggest that strongly coevolving positions of KDO8PS may be associated with one or more of these demands. In order to determine the specific contribution of coevolution to stability we compared the level of MI of each pair with the average contribution to stability by that pair in the MSA of KDO8PS, as derived by applying to each sequence in the MSA the information contained in the stability matrix shown above. For example, based on that matrix a proline would contribute 3.27 kcal/mol at position 31 of a sequence in the MSA, but -1.05 kcal/mol at position 56 (Nm_KDO8PS_energy_matrix). We were interested not only in the sum (DDGi + DDGj) of the stability contributions of each member of the i,j pair (as these contributions can be considered approximately additive under the assumption of no epistasis), but also in the absolute value of the difference |DDGi - DDGj|, which depends on whether the two contributions have similar or opposite effects. Since MI is already the log of a probability, the two magnitudes of interest are:
![]()
![]()
where the average in the two equations is over all the sequences in the MSA. Less than 20% of all coevolving pairs with a score > 5 s in the ZRes matrix have values of |DDGi - DDGj|>(DDGi + DDGj), and thus identify positions that, on average, exert opposite effects on the stability of KDO8PS. For each type of MI matrix, the correlations between the score vector and the (DDGi + DDGj) and |DDGi - DDGj| vectors shows that, as progressively higher s thresholds are chosen, the score vectors become more clearly anti-correlated to the (DDGi + DDGj) vector: this trend is consistent with the two equations above, because we expect that pairs whose components raise the DDG are less likely to occur. The score vectors are also anti-correlated to the |DDGi - DDGj| vector, suggesting that on average coevolving pairs have a preference for positions whose individual effects on stability are similar.
These results can be interpreted in the light of current theories of coevolution. According to one hypothesis, a reason for coevolving pairs is to suppress a decrease in stability or function produced by a mutation at one site with an increase in stability or function provided by a mutation in a residue near the site of the first mutation. This tenet is challenged by the observation that most suppressor mutations are not close in space to the initial mutations, while coevolving sites are most often spatially clustered. Directed evolution studies also suggest that epistatic paths can be bypassed (and they certainly are in laboratory evolution), because there are multiple sequences that satisfy a given fitness goal, and there are many different paths to these sequences.
According to an alternative model for coevolution, “covarions” arise when both the original residue and the mutated residue are compatible with function, but the spectrum of residues possible at other positions in the protein is altered by the mutation. In this context one might expect that most single mutations that finally become stabilized in coevolving pairs are neutral in the context of the protein in which they occur, but become beneficial (even at a much later time) in the presence of the partner in the pair [63]. The data in Table 3 suggest that both mechanisms were operational in the evolution of KDO8PS. Approximately 1/4 of all strongly coevolving pairs (for example those shaded in Table 2; see also Table 3) may have originated from cycles of mutation and suppression that affected stability. Other pairs, for which both values of (DDGi + DDGj) and |DDGi - DDGj| are small, are best explained by a succession of neutral or nearly neutral covarions.
Key papers:
The contribution of coevolving residues to the stability of KDO8P synthaseSH Ackerman, DL Gatti
PloS one 6 (3), e17459