This work is conducted in collaboration with Sharon H. Ackerman.
Class B β-lactamases (MβLs) are metallo-enzymes requiring one or two Zn2+ ions for activity. MβLs are now a worldwide threat, because they hydrolyze all β-lactams with the exception of monobactams, and are not inhibited by serine β-lactamase inhibitors, such as clavulanic acid, tazobactam, and sulbactam. MβLs are particularly efficient as carbapenemases, which is very alarming because carbapenems are the antibiotics with the largest spectrum of activity, and are stable to hydrolysis by most of the serine β-lactamases. MβLs are evolving rapidly and becoming progressively more effective and specialized against different antibiotics; unfortunately, no clinically relevant inhibitors of these enzymes are yet available .
All MβLs possess two potential Zn2+ sites, but differ in Zn2+ occupancies and coordination environments. In B1 enzymes one Zn is coordinated by His116, His118, His196 (Site 1 or Zn1), and the other Zn by Asp120, Cys221, His263 (Site 2 or Zn2). A water molecule or OH- ion bridges the two Zn2+ ions. In B2 enzymes Site 1 is incomplete (having only His118 and His196) and without metal, and only Site 2 is occupied. In B3 enzymes Site 1 is unchanged, while at Site 2 His121 replaces Cys221 as a Zn ligand. B1 and B3 enzymes have maximum activity as di-zinc species. In contrast, B2 enzymes exhibit maximal activity when bound to only one Zn, the binding of a second Zn ion being inhibitory.
A general mechanism has been proposed for MβLs, in which deprotonation of a water molecule at the Zn2 site results in the formation of a hydroxide ion that attacks the carbonyl oxygen of the b-lactam ring. The original Zn-bound water/hydroxide ion is lost in this reaction and is replaced by a new solvent molecule that enters the active site before the next turnover. However, it is not clear that this mechanism applies to all MβLs.
We have adopted a novel strategy to identify the RS of the MβL reaction, which does not rely on substrate docking and/or molecular dynamics. Starting with the X-ray structure of the enzyme:product complex, the protonation states of the product and of key groups in the active site are assigned in such a way that these molecules contain all the atoms expected to be present in the enzyme:substrate complex at the RS. Then, a QM/MM scan is used to drive the reaction uphill from product back to reactant. As the reaction is driven backward also the conformation of the enzyme reverts to that of the reactant state. In so doing we actually generate the enzyme:substrate complex from product and avoid the uncertainties associated with modeling the RS. Using this strategy, we studied the reaction of biapenem hydrolysis by CphA from Aeromonas hydrophila, a B2 type MβL.
The structure of N220G CphA is available (PDB entry 1X8I) in complex with a form of bicyclic hydrolyzed biapenem that has undergone a molecular rearrangement such that oxygen atom O62 forms a 6-membered ring (N4-C5-C6-C61-O62-C3) that replaces the original β-lactam ring. Since the bicyclic compound is a rearranged form of the product of β-lactam ring hydrolysis, starting from the structure in PDB entry 1X8I, in our previous work on CphA using a combination of QM/MM and metadynamics simulations we generated a form of the enzyme:product complex in which the steps of the post-hydrolysis reactions have been reversed. This modified structure served as the starting point for the application of a series of 2-dimensional QM/MM scans to generate 14 different potential energy surfaces (PESs) corresponding to a large variety of possible reaction paths. Only some of these PESs (corresponding to mechanisms in which the barrier is similar to the barrier determined experimentally,~14 kcal/mol) are described here.
Active site configuration 2.
In this simulation, at the PS with the lowest energy biapenem N4 is deprotonated, Wat2 is not ionized, Asp120 is deprotonated, and His118, and His196 are neutral. The CBIA–OBIA/WAT1, HNBIA–NBIA distances were used as the scanning coordinates in the construction of the PES (the latter is the H–N bond that is formed when the amide nitrogen of the product is protonated). In this case, a barrier of ~15 kcal/mol separates the reactant from the product: this barrier corresponds to an attack by WAT1 on the carbonyl carbon (C7) of the lactam ring. Past the TS, the reaction leads to either a protonated or unprotonated product. However, the energy level of the unprotonated product is ~10 kcal/mol lower than that of protonated product and a barrier of ~20 kcal/mol (labeled 4) separates the two states.
Active site configuration 3.
A very similar configuration of the active site was explored in a different simulation, the only difference with respect to Simulation 2 being that Asp120 was protonated in the product state with the lowest energy. In this case a barrier of 12 kcal/mol separates the RS from the unprotonated product. Formation of a protonated product occurs over a much higher barrier of ~25 kcal/mol.
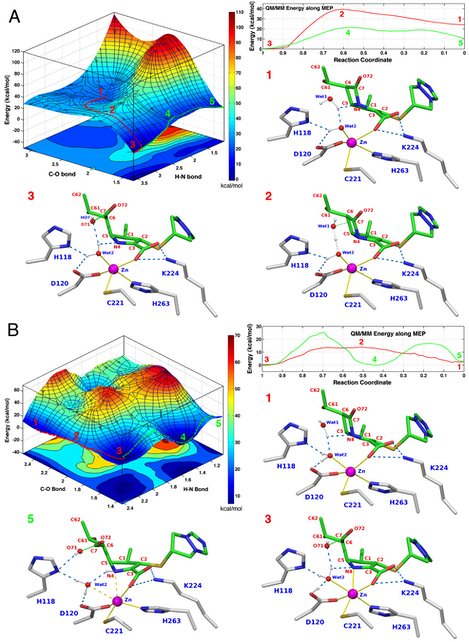
A. Top left. PES of the reaction calculated using the C–O and H–N bonds as scanning coordinates. Two possible product states are visible on the PES corresponding respectively to hydrolyzed biapenem with unprotonated (labeled 3) or protonated N4 (labeled 5). The minimum energy path (MEP) from RS to unprotonated PS is traced by a red string. The MEP from this PS to the PS in which N4 is protonated is shown as a green string. Top right. QM/MM energy values along the two MEPs. RS and ionized PS are separated by a barrier of ~15 kcal/mol, and the reaction is strongly exergonic (-24 kcal/mol). Protonated product is ~10 kcal/mol higher in energy than unprotonated product and a barrier of ~20 kcal/mol separates the two states. The other three insets show the configurations of the active site corresponding to the RS, TS, and PS along the red MEP on the PES. At the RS (inset 1) Wat2 is present as a hydroxide ion. Coincident with the formation of a tetrahedral TS, a proton is shared between Wat1 and the hydroxide ion (inset 2). Concurrent with the opening of the ring (inset 3) the proton is transferred to Wat2. Thus, the reaction proceeds through the formation of a tetrahedral TS, and the rate-limiting step is the concurrent formation of the C–O bond while the C–N bond is broken. There is no proton transfer to His118, His196 or Asp120.
B. Top left. PES of the reaction calculated using the C–O and H–N bonds as scanning coordinates. Two possible product states are visible on the PES corresponding respectively to hydrolyzed biapenem with unprotonated or protonated N4. The MEP from RS to unprotonated PS is traced by a red string. The MEP from this PS to the PS in which N4 is protonated is shown as a green string. Top right. QM/MM energy values along the two MEPs. RS (labeled 1) and ionized PS (labeled 3) are separated by a barrier of ~12 kcal/mol, and the reaction is only slightly exergonic (-3 kcal/mol). Protonated product (labeled 5) is isoenergetic with unprotonated product, but a barrier of ~25 kcal/mol separates the two states. The other three insets show the configurations of the active site corresponding to the RS, ionized PS, and protonated PS. At the RS (inset labeled 1) both Wat1 and Wat2 are fully protonated. At the ionized PS a proton has been transferred from Wat1 to Wat2, and from Wat2 to Asp120 (inset labeled 3): thus Wat2 remains fully protonated. At the protonated PS (inset labeled 5) a proton has been transferred from Wat2 to biapenem N4, and from Asp120 to Wat2: thus also in this case Wat2 remains fully protonated. The most favorable reaction proceeds through the formation of a tetrahedral TS (labeled 2 on the PES), and the rate-limiting step is the concurrent formation of the C–O bond and the breaking of the C–N bond.
A complete vibrational analysis of the transition (TS) and stationary points (SP) on the PSE derived from simulation 3 (which has the lowest overall energy barrier for the hydrolysis step) was carried out to convert the values of electronic energy into free-energy values. The free energy profile of the reaction so derived is qualitatively similar to the electronic energy profile shown above, and reveals again the largest barrier to be the step of N4 protonation.
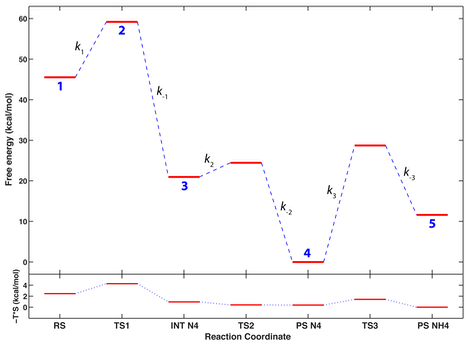
Upper quadrant. Stationary and TS points are represented as red thick horizontal lines connected by dashed blue lines; numbers in blue (1-5) below each red line correspond to the same numbers on the PES. The reaction coordinate axis is in arbitrary units and the stationary points are marked as follows: RS is biapenem; INT N4 is an intermediate conformation of the active site in which the b-lactam ring of biapenem is already open and N4 is ionized; PS N4 is a slightly changed conformation of the active site in which Wat2 becomes closer to hydrolyzed biapenem, but N4 is still ionized; PS NH4 is the open-ring form of biapenem with N4 protonated. Lower quadrant. Changes in the entropic contribution (-T*S) to the free energy profile shown in the upper quadrant. Both the free energy and the entropy profile are not on an absolute scale, but were shifted such that their smallest value would correspond to 0 on the energy axis.
The rate constants derived from simulation 3 for the steps of the hydrolysis reaction can be combined with those determined in our previous study for the steps of the post-hydrolysis conversion of biapenem to a bicyclic product to generate a complete kinetic model of biapenem inactivation by CphA. A version of this model including all the kinetic laws is provided below in Matlab SimBiology format (CphA_Kinetic_Model_S1) and SBML format (CphA_Kinetic_Model_S2). The model includes two symmetric branches for the generation of the bicyclic derivative of Biapenem both inside the enzyme (green species in figure below) and in the solvent bulk-phase (cyan species in figure below). This model can be conveniently used in future studies for further refinement of the kinetic parameters of the reaction of biapenem inactivation by CphA against both steady-state and pre-steady-state experimental observations.
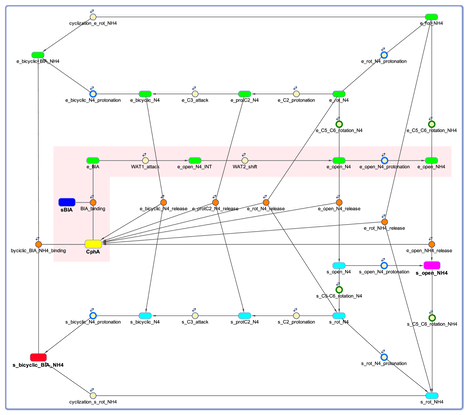
The model describes the binding of biapenem (blue box) to CphA (yellow box) and its subsequent conversion to an open-ring form (purple box) and to a bicyclic product (red box). Formation of the bicyclic derivative of biapenem occurs both in the enzyme (species labeled ‘e_’) and in solution (species labeled ‘s_’). All other enzyme-bound and bulk-solvent species are represented as rounded green and cyan boxes, respectively. Species with N4 ionized are labeled ‘_N4’, while those with N4 protonated are labeled ‘_NH4’. Reactions are shown as circles with arrows connecting the species involved. Orange-filled circles represent binding/release reactions; green-outlined circles represent rotations of the hydroxyethyl moiety around the C5-C6 bond; blue-outlined circles represent protonations of the N4 nitrogen. A pink shaded area highlights the reaction corresponding to Simulation 3. Parameters for all the other reactions were derived from our earlier studies.
Kinetic models:
Key papers:
Biapenem Inactivation by B2 Metallo β-Lactamases: Energy Landscape of the Post-Hydrolysis Reactions
DL Gatti
PloS one 7 (1), e30079
Biapenem Inactivation by B2 Metallo β-Lactamases: Energy Landscape of the Hydrolysis Reaction
SH Ackerman, DL Gatti
PloS one 8 (1), e55136