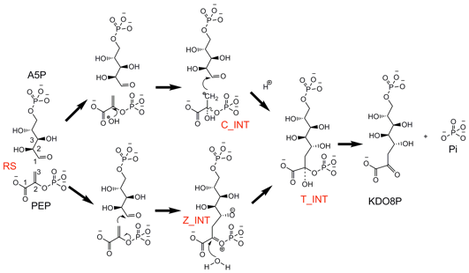
Two stepwise mechanisms have been proposed for the condensation reaction of PEP and A5P to form KDO8P and Pi. One mechanism involves a transient oxocarbenium zwitterionic intermediate (Z_INT), formed by direct attack of C3PEP onto C1A5P, followed by reaction of water at C2Z_INT (lower path in the figure below). This mechanism was originally proposed for non-metallo KDO8PSs, but then extended also to the metal forms, in which it was thought that the metal ion might play a role as a Lewis acid by coordinating the aldehyde group of A5P. A different mechanism was proposed for metallo KDO8PSs (upper path in the figure below), which involved a transient carbanion intermediate (C_INT), formed by attack of water or a hydroxide ion on C2PEP. This mechanism relied on the assumption that the metal would favor the deprotonation of the coordinated water to a hydroxide ion, which would be a good nucleophile for attack on C2PEP. The ensuing carbanion at C3PEP would then facilitate attack of C3PEP to C1A5P. In both mechanisms, the transient intermediates (C_INT or Z_INT) are expected to converge to a more stable tetrahedral intermediate (T_INT), which decays into KDO8P and Pi. T_INT has been observed in the crystal structure of the C11N/S235P/Q237A triple mutant of A. aeolicus KDO8PS (see X-ray crystallography page).
The configuration of the C2T_INT chiral center in one of the active sites visible in this structure suggests that water attacks the si side of C2PEP or C2Z_INT such that the overall condensation reaction would correspond to a syn addition of water and A5P to the C2 and C3 carbons of PEP. However, in the other active sites of the same crystal structure a water molecule is not visible on the si side of PEP when A5P is also bound, making it difficult to explain how the syn addition might have occurred. Furthermore, in the crystal structures of all KDO8PSs there is another water molecule on the re side of C2PEP, which would lead to the arguably more stable anti addition of water and A5P to the C2-C3 double bond of PEP.
We have explored the reaction of KDO8P synthesis in the C11N form of A. aeolicus KDO8PS (which under all respects can be considered as a non-metal variant of the A. aeolicus enzyme) with a systematic construction by quantum mechanics/molecular mechanics (QM/MM) calculations of the potential energy surfaces (PESs) corresponding to all possible reaction paths.
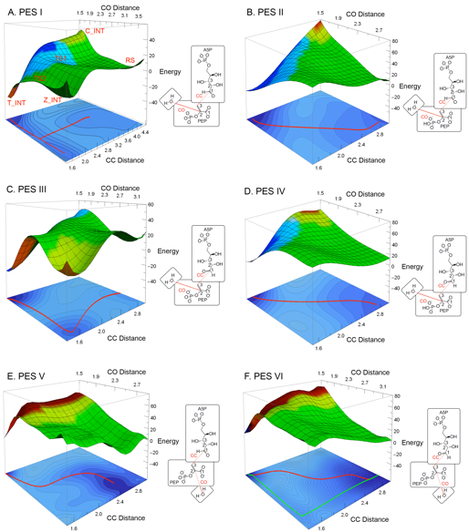
Potential Energy Surface of KDO8P synthesis. A. PES I, reaction in which OWAT attacks the si side of C2PEP and C3PEP attacks the re side of C1A5P: PEP phosphate is deprotonated. B. PES II, same as A, but with PEP phosphate partially protonated. C. PES III, reaction in which OWAT attacks the si side of C2PEP and C3PEP attacks the si side of C1A5P: PEP phosphate is deprotonated. D. PES IV, same as C, but with PEP phosphate partially protonated. E. PES V, reaction in which OWAT attacks the re side of C2PEP and C3PEP attacks the re side of C1A5P: PEP phosphate is deprotonated. F. PES VI, reaction in which OWAT attacks the re side of C2PEP and C3PEP attacks the si side of C1A5P: PEP phosphate is deprotonated. CC and CO distances are in Å, energies are in kcal/mol. Colors on the PESs represent in a qualitative way proton movements along the reaction path: green for proton on OWAT, blue for proton nearing O1A5P, red for proton nearing PEP phosphate oxygen. Colors on the projected contours below the PESs reflect energy levels, with darker shades of blue corresponding to lower energy.
Among the various PESs calculated in this study, the lowest energy reaction path from RS (PEP + A5P + H2O) to T_INT (the experimentally observed intermediate) was observed on PES I, which describes a syn addition of water to the si side of C2PEP and of C3PEP to the re side of C1A5P when the phosphate group of PEP is deprotonated. The calculated barrier and reaction energy for this path are ~14 kcal/mol and –38 kcal/mol, respectively. Other possible paths, like an anti addition of water to the re side of C2PEP or addition of C3PEP to the si side of C1A5P are associated with much higher energy barriers (PESs III, V, VI). These results are consistent with established features of the reaction, and thus confirm the validity of the calculations. The new finding of this study is that the lowest energy path to the formation of T_INT does not correspond to either of the pure stepwise processes described above, but corresponds instead to a partially concerted reaction between PEP, A5P, and water in the enzyme active site.
In trying to understand the role of metal in KDO8PS it is useful to consider whether Lenhinger’s principle applies: if the catalytic activity is already present in the metal alone, the enzyme will likely act by amplifying that intrinsic activity. Synthesis of KDO8P was reported by Baasov’s group to occur in an aqueous solution at pH 5.0 with a t1/2 ≈ 5hrs in the presence of Zn2+ by intramolecular reaction of the double bond of the enolpyruvyl moiety with the aldehyde moiety in a model compound synthesized by tethering an enolpyruvyl functionality to the A5P C3-OH functionality [45].
In this reaction the metal was believed to act as a Lewis acid by activating the aldehyde carbonyl. Unfortunately, non-enzymatic synthesis of KDO8P in water from PEP and A5P, which is the exact counterpart of the reaction occurring in the enzyme, has not been studied, as this reaction is believed to be too slow or not to occur at all in the absence of KDO8PS. However, we were able to explore this reaction by QM/MM . The methods used in this study were similar to those used in our QM/MM simulations of both metallo and non-metallo KDO8PSs described above. The PES for the non-enzymatic condensation of PEP and A5P occurring inside a sphere of water molecules of 26 Å radius was defined by two reaction coordinates: the formation of the bond between C3PEP and C1A5P, and the formation of the bond between the oxygen of water and C2PEP. PESs were calculated in the absence or presence of Zn2+, and in the latter case, with Zn2+ coordinated in the reactant state (RS) to the water molecule that attacks C2PEP or to the aldehyde carbonyl oxygen of A5P. In all cases the minimum energy path on the PES leads first to a C3PEP–C1A5P bond, and then to an OWAT–C2Z_ION bond, suggesting that in the non-enzymatic condensation of PEP and A5P, Z_ION is formed first (1st transition state, TS1), and then water reacts with it (2nd transition state, TS2): in all cases, formation of Z_ION appears to be the rate limiting step.
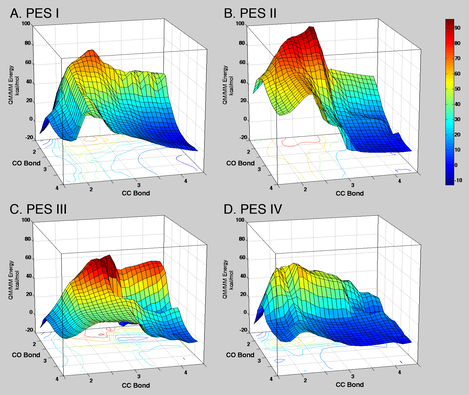
PESs
of the non-enzymatic condensation of PEP and A5P in water.
The reactants (PEP + A5P + WAT with/without Zn2+/acetamide) were
immersed in a sphere of explicit waters of 26 Å radius. A5P, PEP, the single
water molecule involved in the reaction, and the metal ion or the acetamide
molecule were treated quantum mechanically. All the other water molecules were treated with
a molecular mechanics force field. Each PES is
defined by two reaction coordinates: formation of the bond between C3PEP
and C1A5P, and formation of the bond between the oxygen of water and
C2PEP. A. PES I: no Zn2+ or acetamide. B. PES II: at RS
Zn2+ is coordinated to the water molecule that attacks C2PEP;
at TS1 and TS2 Zn2+ is coordinated to the same water molecule and to
A5P carbonyl oxygen. C. PES III: Zn2+ is coordinated to A5P carbonyl
oxygen. D. PES IV: acetamide (mimicking an asparagine side chain) is hydrogen
bonded to A5P carbonyl oxygen. CC and CO distances are in Å, QM/MM energies are
in kcal/mol. Colors on the PESs and on the projected contours below the PESs
reflect QM/MM energy levels, as represented in the reference bar on the side.
Without metal the simulated reaction (PES I) is exothermic (DH = -11 kcal/mol) with an overall barrier on the minimum energy path of ~24 kcal/mol. In the reaction corresponding to PES II at the RS Zn2+ is coordinated only by the water molecule that attacks PEP, while at TS1 and TS2 it is coordinated also by the carboxylate moiety of PEP and the carbonyl oxygen of A5P . The overall reaction is endothermic (DH = + 32.0 kcal/mol) with a barrier of 35.0 kcal/mol. When Zn2+ is coordinated only to the carbonyl oxygen of A5P throughout the entire simulation (PES III), the reaction is exothermic (DH = -14 kcal/mol) with a barrier of ~22 kcal/mol (Table 1). Overall, the shape of the PES is somewhat unusual, with an extended plateau corresponding to the progressive formation of the C3PEP–C1A5P bond. Despite the lack of a clear saddle point in the PES a transition state search and the vibrational analysis identify unambiguously a single TS1, characterized by a rather large C3PEP to C1A5P distance (2.6 Å). The reaction corresponding to PES IV was studied to simulate the effect of an asparagine side chain hydrogen bonded to the carbonyl oxygen of A5P. The rationale for this investigation lies in the fact that in the active site of non-metallo KDO8PSs an asparagine side chain replaces the metal ion bound to a cysteine side chain (see above). Furthermore, a hydrogen bond between the amine moiety of Asn11 and the carbonyl oxygen of A5P or the C4–OH of INT (which originates from the carbonyl oxygen of A5P) was observed in two metal independent forms of Aa. KDO8PS carrying the C11N substitution. We have gained some insight into the role of this asparagine side chain in the enzymatic reaction, by computing the PES of the non-enzymatic reaction in water when the amine moiety of the model compound acetamide (which mimics an asparagine side chain) is hydrogen bonded to the carbonyl oxygen of A5P (PES IV). The simulated reaction is exothermic (DH = -11 kcal/mol) with a first barrier of 16.9 kcal/mol corresponding to the formation of a C–C bond between C3PEP and C1A5P (TS1), and a second barrier of 5.0 kcal/mol corresponding to the formation of a C–O bond between water and Z_ION (TS2). In both TSs C1–OA5P or C4–OZ_ION are hydrogen bonded to the amine moiety of acetamide. An important feature of TS1 is the orientation of the C2PEPPEP–C3 bond with respect to the C1A5P–C2A5P bond. These bonds are almost perpendicular to each other at TS1 in PESs I, II, and III, but perfectly co-linear at TS1 in PES IV.
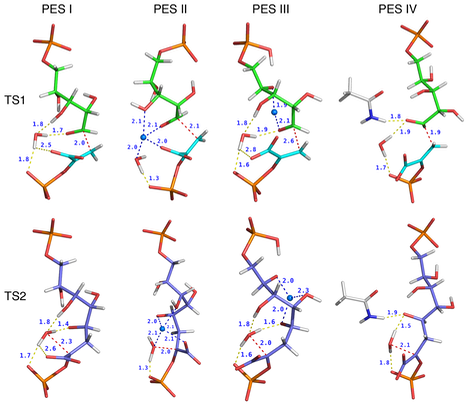
Transition states in the non-enzymatic condensation of PEP and A5P in water. The relative positions of PEP, A5P, Z_ION, Zn2+, water, and acetamide at TS1 (top row) and TS2 (bottom row) are shown for the PESs (I to IV) . PEP is shown with cyan bonds, A5P with green bonds, Z_ION with purple bonds, acetamide with white bonds. Zn2+ is shown as a light-blue sphere. Hydrogen bonds are shown as yellow dashed lines. Zn2+ coordination is shown as blue dashed lines. Red dashed lines represent the C3PEP–C1A5P bond (TS1) or the OWAT–C2Z_ION bond (TS2) and reflect a pure bond-stretching mode corresponding to the single imaginary frequency identified at TS1 and TS2. Distances in Angstroms are shown next to each line.
Thus, the PES of the reaction in the active site of KDO8PS (see above) is similar to that computed for the same reaction inside a sphere of water molecules, without enzyme and with added acetamide. The non-enzymatic reaction is predicted to be clearly step-wise with formation of a transient oxocarbenium ion (Z_ION), while the enzymatic reaction is predicted to have at least a partial character of a concerted reaction between PEP, A5P, and water. In both cases, formation of the C3PEPA5P–C1 bond represents the bottleneck of the reaction.
In conclusion, QM/MM studies of KDO8P synthesis either in solution, with key chemical groups mimicking the catalytic side chains, or with a complete simulation of the non-metallo enzyme active site suggest that a role equivalent to that of the metal is performed by an asparagine side chain that in the non-metallo form of the enzyme replaces the combination cysteine + metal. While a complete simulation of the reaction in the metallo enzyme is not available yet, the QM/MM optimized geometries of the RS and INT states for both enzyme forms suggest that the active site metal or the amine moiety of Asn11 stimulate the reaction by positioning A5P and water in a similar way with respect to PEP in both metallo and non-metallo KDO8PSs.
The evolution of almost identical metal dependent and metal-independent forms of KDO8PS was possible because the metal does not appear to be directly involved in an activation process, but primarily contributes to stabilize the relative positions and conformations of the reactants that allow for the lowest activation energy. Thus, if a general conclusion can be drawn from these studies, it would be that, following Lehninger’s original suggestion, attempts to convert metallo-enzymes to non-metallo forms or vice versa should focus on clarifying whether the metal has a direct activating role or just contributes to orient the substrates in the active site. The accumulated results from mutagenesis, crystallographic and computational studies of KDO8PS suggest that a minimalist strategy to achieve this conversion is more likely to succeed in the latter case.
Key papers:
Electronic
Structure of the Metal Center in the Cd [superscript 2+], Zn
[superscript 2+], and Cu [superscript 2+] Substituted Forms of KDO8P
Synthase: Implications for Catalysis
F Kona, P Tao, P Martin, X Xu, DL Gatti
Biochemistry-US 48 (16); 04, 2009)
The energy landscape of 3-deoxy-D-manno-octulosonate 8-phosphate synthase
P Tao, DL Gatti, HB Schlegel
Biochemistry 48 (49), 11706-11714
Common basis for the mechanism of metallo and non-metallo KDO8P synthases
P Tao, HB Schlegel, DL Gatti
Journal of inorganic biochemistry 104 (12), 1267-1275